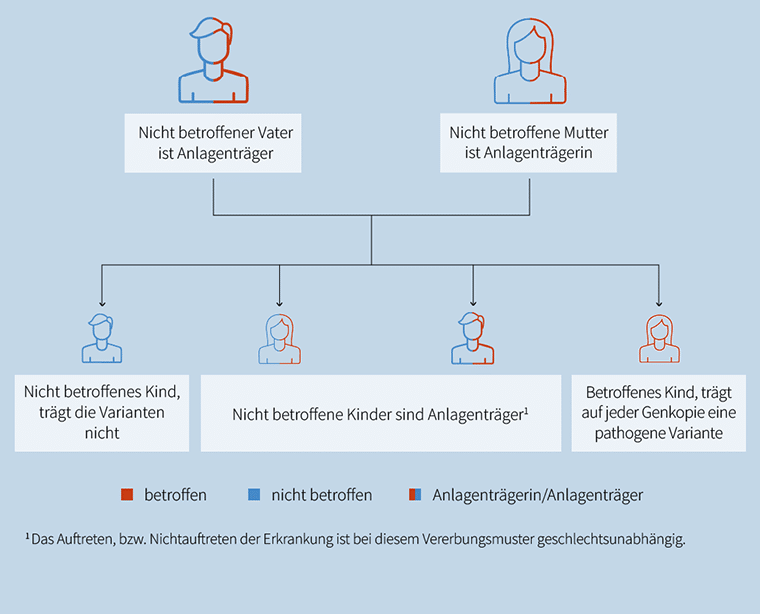
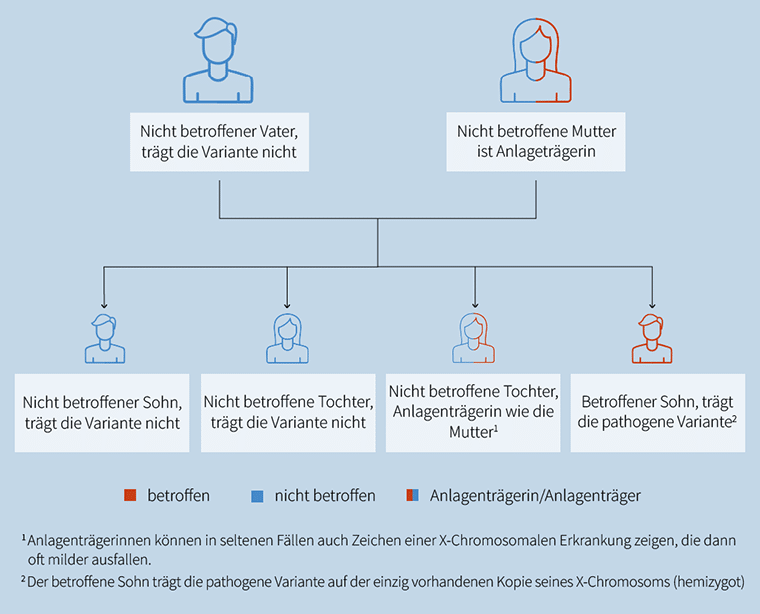
AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADSB, ACADVL, ACAN, ACAT1, ACD, ACE, ACO2, ACOX1, ACOX2, ACP5, ACSL4, ACTA1, ACTL6B, ACY1, ADA, ADA2, ADAM17, ADAM22, ADAMTS13, ADAMTS19, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY1, ADCY5, ADCY6, ADGRG1, ADGRG6, ADGRV1, ADK, ADPRS, ADSL, AFF2, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHI1, AIFM1, AIMP1, AIMP2, AIPL1, AIRE, AK2, AKR1D1, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALOXE3, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK3, ANKLE2, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP3D1, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AQP2, AR, ARFGEF2, ARG1, ARHGDIA, ARHGEF9, ARL13B, ARL3, ARL6, ARL6IP1, ARMC9, ARNT2, ARPC1B, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ATAD1, ATAD3A, ATCAY, ATIC, ATM, ATOH7, ATP13A2, ATP1A2, ATP2B3, ATP5F1D, ATP5MK, ATP6AP1, ATP6AP2, ATP6V0A2, ATP6V0A4, ATP6V1A, ATP6V1B1, ATP6V1E1, ATP7A, ATP7B, ATP8A2, ATP8B1, ATPAF2, ATR, ATRX, AUH, AVIL, B3GALNT2, B3GALT6, B3GAT3, B3GLCT, B4GALNT1, B4GALT1, B4GALT7, B4GAT1, B9D1, B9D2, BANF1, BBIP1, BBS1, BBS10, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, BCAP31, BCKDHA, BCKDHB, BCKDK, BCOR, BCS1L, BGN, BHLHA9, BIN1, BLM, BLNK, BLTP1, BMP1, BMP2, BMPER, BMPR1B, BOLA3, BPNT2, BRAT1, BRCA1, BRCA2, BRF1, BRWD3, BSCL2, BSND, BTD, BTK, BUB1B, C12orf57, C19orf12, C1QBP, C2CD3, C2orf69, CA2, CA5A, CA8, CABP2, CACNA1D, CAD, CAMK2A, CANT1, CAPN3, CARD11, CARMIL2, CARS2, CASK, CASQ2, CASR, CAV1, CAVIN1, CBS, CC2D1A, CC2D2A, CCBE1, CCDC103, CCDC115, CCDC22, CCDC39, CCDC40, CCDC47, CCDC65, CCDC8, CCDC88A, CCDC88C, CCN6, CCNO, CCNQ, CCT5, CD19, CD247, CD27, CD3D, CD3E, CD3G, CD40, CD40LG, CD55, CD70, CD79A, CD79B, CDC14A, CDC45, CDH11, CDH2, CDH23, CDH3, CDIN1, CDK10, CDK5RAP2, CDKL5, CDSN, CDT1, CENPF, CENPJ, CEP104, CEP120, CEP135, CEP152, CEP164, CEP290, CEP41, CEP55, CEP57, CEP63, CEP78, CEP83, CERS1, CERS3, CFAP298, CFAP300, CFAP410, CFAP418, CFL2, CFP, CFTR, CHAT, CHKB, CHM, CHMP1A, CHRDL1, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, CHST14, CHST3, CHSY1, CHUK, CIB2, CIITA, CILK1, CISD2, CIT, CKAP2L, CLCN1, CLCN2, CLCN4, CLCN5, CLCN7, CLCNKB, CLDN1, CLDN10, CLDN14, CLDN16, CLDN19, CLIC5, CLMP, CLN3, CLN5, CLN6, CLN8, CLP1, CLPB, CLPP, CLRN1, CNKSR2, CNNM2, CNPY3, CNTNAP1, CNTNAP2, COA6, COA8, COASY, COCH, COG1, COG2, COG4, COG5, COG6, COG7, COL11A1, COL11A2, COL13A1, COL17A1, COL18A1, COL1A2, COL27A1, COL3A1, COL4A3, COL4A4, COL4A5, COL6A1, COL6A2, COL6A3, COL7A1, COL9A2, COLEC10, COLEC11, COLQ, COQ2, COQ4, COQ6, COQ7, COQ8A, COQ8B, COQ9, CORO1A, COX10, COX14, COX15, COX20, COX6A2, COX6B1, COX7B, COX8A, CPLANE1, CPLX1, CPS1, CPT1A, CPT2, CRADD, CRB1, CRB2, CRBN, CREB3L1, CRIPT, CRLF1, CRPPA, CRTAP, CRYAA, CRYAB, CSF1R, CSF2RB, CSF3R, CSPP1, CSTA, CSTB, CTC1, CTDP1, CTNNA2, CTNS, CTPS1, CTSA, CTSD, CTSK, CTU2, CUL4B, CUL7, CWC27, CWF19L1, CYB5R3, CYBA, CYBB, CYC1, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP24A1, CYP27A1, CYP27B1, CYP2R1, CYP2U1, CYP4F22, CYP7B1, D2HGDH, DAG1, DARS1, DARS2, DBT, DCAF17, DCDC2, DCHS1, DCLRE1C, DCX, DDB2, DDC, DDHD1, DDHD2, DDR2, DDX11, DDX3X, DDX59, DEAF1, DEGS1, DENND5A, DGAT1, DGKE, DGUOK, DHCR24, DHCR7, DHDDS, DHH, DHODH, DHTKD1, DHX37, DIAPH1, DIS3L2, DKC1, DLAT, DLD, DLG3, DLL3, DLX5, DMD, DMP1, DMXL2, DNA2, DNAAF11, DNAAF3, DNAAF4, DNAAF5, DNAAF6, DNAH11, DNAH5, DNAH9, DNAJC12, DNAJC19, DNAJC21, DNAJC3, DNAJC6, DNM1L, DNM2, DNMT3B, DOCK2, DOCK6, DOCK7, DOCK8, DOK7, DOLK, DONSON, DPAGT1, DPH1, DPM1, DPM2, DPYD, DRC1, DSE, DSG1, DSP, DST, DSTYK, DUOX2, DUOXA2, DYM, DYNC2H1, DYNC2I1, DYNC2I2, DYNC2LI1, DYSF, EARS2, EBP, ECEL1, ECHS1, EDA, EDAR, EDARADD, EDN3, EDNRB, EFEMP2, EFL1, EFNB1, EGR2, EIF2AK3, EIF2AK4, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S3, EIF4A3, ELAC2, ELMO2, ELMOD3, ELOVL4, ELP1, ELP2, EMC1, EMC10, EMD, EMG1, EML1, ENPP1, ENTPD1, EOGT, EPCAM, EPG5, EPM2A, EPRS1, EPS8, EPS8L2, ERAL1, ERBB3, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, ERCC6, ERCC6L2, ERCC8, ERLIN1, ERLIN2, ESCO2, ESPN, ESRRB, ETFA, ETFB, ETFDH, ETHE1, EVC, EVC2, EXOC3L2, EXOSC3, EXOSC8, EXOSC9, EXPH5, EXT2, EXTL3, F10, F13A1, F2, F7, F8, F9, FA2H, FADD, FAH, FAM126A, FAM149B1, FAM20A, FAM20C, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, FAR1, FARS2, FASTKD2, FAT4, FBLN5, FBP1, FBXL4, FBXO7, FCSK, FERMT3, FEZF1, FGA, FGB, FGD1, FGD4, FGF3, FGFR3, FGG, FH, FHL1, FIG4, FITM2, FKBP10, FKBP14, FKRP, FKTN, FLAD1, FLNA, FLNB, FLVCR1, FLVCR2, FOLR1, FOXE1, FOXE3, FOXL2, FOXN1, FOXP3, FOXRED1, FRAS1, FREM1, FREM2, FRMPD4, FRRS1L, FSHB, FTCD, FTL, FTO, FTSJ1, FUCA1, FUT8, FXN, G6PC1, G6PC3, GAA, GAD1, GALC, GALE, GALK1, GALNS, GALT, GAMT, GAN, GAS8, GATA1, GATM, GBA1, GBA2, GBE1, GCDH, GCH1, GCK, GCSH, GDAP1, GDF1, GDF5, GDF6, GDI1, GEMIN4, GFER, GFM1, GFM2, GFPT1, GHR, GIPC3, GJA1, GJB2, GJB3, GJB6, GJC2, GK, GLA, GLB1, GLDC, GLDN, GLE1, GLIS3, GLRX5, GLS, GLUL, GLYCTK, GM2A, GMPPA, GMPPB, GNB5, GNPAT, GNPTAB, GNPTG, GNRH1, GNRHR, GNS, GOLGA2, GORAB, GOSR2, GOT2, GPAA1, GPC3, GPC6, GPHN, GPSM2, GPT2, GPX4, GRHL2, GRHPR, GRIA3, GRID2, GRIK2, GRIN1, GRIP1, GRM1, GRM7, GRXCR1, GSS, GTF2H5, GTPBP3, GUCY1A1, GUCY2C, GUF1, GUSB, GYS1, GYS2, GZF1, HACD1, HACE1, HADH, HADHA, HADHB, HAMP, HARS1, HARS2, HAX1, HBB, HCCS, HCFC1, HDAC8, HEPACAM, HERC1, HERC2, HES7, HESX1, HEXA, HEXB, HFE, HGF, HGSNAT, HIBCH, HIKESHI, HINT1, HJV, HK1, HLCS, HMGCL, HMGCS2, HMX1, HNRNPH2, HOGA1, HOXA1, HOXC13, HPD, HPDL, HPGD, HPRT1, HPS1, HPSE2, HSD11B2, HSD17B10, HSD17B3, HSD17B4, HSD3B2, HSD3B7, HSPA9, HSPD1, HSPG2, HTRA2, HUWE1, HYAL1, HYDIN, HYLS1, IARS1, IARS2, IBA57, ICOS, IDS, IDUA, IER3IP1, IFIH1, IFNGR1, IFNGR2, IFT122, IFT140, IFT172, IFT27, IFT43, IFT52, IFT74, IFT80, IFT81, IGBP1, IGF1, IGF1R, IGFBP7, IGHMBP2, IGSF1, IHH, IKBKB, IKBKG, IL10RA, IL11RA, IL12RB1, IL1RAPL1, IL1RN, IL21R, IL2RA, IL2RB, IL2RG, IL7R, ILDR1, INPP5E, INPP5K, INPPL1, INS, INSR, INTU, INVS, IPO8, IQCB1, IQSEC1, IQSEC2, IRAK4, IRF8, IRX5, ISCA1, ISCA2, ITCH, ITGA3, ITGA6, ITGA7, ITGA8, ITGB4, ITK, ITPA, ITPR1, IVD, JAGN1, JAK3, JAM2, JAM3, JUP, KARS1, KATNB1, KATNIP, KCNE1, KCNJ1, KCNJ10, KCNJ11, KCNMA1, KCNQ1, KCTD7, KDELR2, KDM5B, KDM5C, KDM6A, KIAA0586, KIAA0753, KIDINS220, KIF14, KIF1A, KIF1C, KIF7, KIFBP, KISS1R, KLHL15, KLHL40, KLHL41, KLHL7, KNL1, KPTN, KRT10, KRT14, KRT18, KRT5, KRT8, KY, L1CAM, L2HGDH, LAGE3, LAMA1, LAMA2, LAMA3, LAMB1, LAMB2, LAMB3, LAMC2, LAMC3, LAMP2, LARGE1, LARP7, LARS2, LAS1L, LAT, LBR, LDHA, LDLR, LFNG, LGI4, LHB, LHFPL5, LHX3, LIAS, LIFR, LIG4, LIMS2, LINS1, LIPA, LIPT1, LMBR1, LMBRD1, LMNA, LMOD3, LNPK, LONP1, LOXHD1, LPIN1, LPIN2, LPL, LRBA, LRP2, LRP4, LRP5, LRPPRC, LRRC56, LRTOMT, LTBP2, LTBP3, LTBP4, LYRM4, LYRM7, LYST, LZTFL1, LZTR1, MAB21L2, MAG, MAGI2, MAGT1, MALT1, MAMLD1, MAN1B1, MAN2B1, MANBA, MAOA, MAP3K20, MAPKBP1, MARS1, MARVELD2, MASP1, MAT1A, MATN3, MBOAT7, MBTPS2, MC2R, MCCC1, MCCC2, MCEE, MCM4, MCOLN1, MCPH1, MDH2, MECP2, MECR, MED12, MED17, MED23, MED25, MEFV, MEGF10, MEGF8, MEOX1, MESD, MESP2, MET, METTL23, METTL5, MFN2, MFRP, MFSD2A, MFSD8, MGAT2, MGME1, MGP, MICOS13, MICU1, MID1, MIPEP, MITF, MKKS, MKS1, MLC1, MLPH, MLYCD, MMAA, MMAB, MMACHC, MMADHC, MMP13, MMP2, MMP21, MMUT, MOCS1, MOCS2, MOGS, MPDU1, MPDZ, MPI, MPL, MPLKIP, MPV17, MPZ, MPZL2, MRE11, MRPL3, MRPL44, MRPS14, MRPS16, MRPS2, MRPS22, MRPS34, MSL3, MSMO1, MSN, MSRB3, MSTO1, MTFMT, MTHFD1, MTHFR, MTM1, MTMR2, MTO1, MTR, MTRFR, MTRR, MTTP, MUSK, MUTYH, MVK, MYBPC1, MYBPC3, MYD88, MYH11, MYH3, MYH7, MYL3, MYMK, MYO15A, MYO18B, MYO3A, MYO5A, MYO5B, MYO6, MYO7A, MYO9A, MYOD1, MYPN, MYSM1, NAA10, NADSYN1, NAGA, NAGLU, NAGS, NALCN, NANS, NARS1, NARS2, NAXD, NAXE, NBAS, NBN, NCAPD3, NCF1, NCF2, NCF4, NCKAP1L, NDE1, NDP, NDRG1, NDST1, NDUFA1, NDUFA10, NDUFA11, NDUFA12, NDUFA13, NDUFA2, NDUFA6, NDUFA9, NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, NDUFAF8, NDUFB3, NDUFB8, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NEB, NECAP1, NECTIN1, NECTIN4, NEK1, NEK8, NEK9, NEMF, NEU1, NEUROG3, NEXMIF, NFASC, NFU1, NGF, NGLY1, NHEJ1, NHLRC1, NHP2, NHS, NIPAL4, NKAP, NKX3-2, NKX6-2, NMNAT1, NNT, NODAL, NONO, NOP10, NPC1, NPC2, NPHP1, NPHP3, NPHP4, NPHS1, NPHS2, NPR2, NR0B1, NR1H4, NRROS, NRXN1, NSDHL, NSMCE2, NSMCE3, NSUN2, NT5C2, NT5C3A, NTNG2, NTRK1, NUBPL, NUDT2, NUP107, NUP133, NUP188, NUP62, NUP88, NUP93, NYX, OBSL1, OCLN, OCRL, ODAD1, ODAD2, OFD1, OGDH, OPA1, OPA3, OPHN1, ORAI1, ORC1, ORC4, ORC6, OSGEP, OSTM1, OTC, OTOA, OTOF, OTOG, OTOGL, OTUD5, OTUD6B, OTULIN, OXCT1, OXR1, P3H1, PAH, PAK3, PAM16, PANK2, PAPSS2, PARN, PARS2, PAX3, PC, PCBD1, PCCA, PCCB, PCDH12, PCDH15, PCDH19, PCK1, PCNT, PCSK1, PCYT1A, PCYT2, PDE10A, PDE6D, PDE6G, PDHA1, PDHB, PDHX, PDP1, PDSS1, PDSS2, PDZD7, PEPD, PERCC1, PET100, PEX1, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6, PEX7, PFKM, PGAP1, PGAP2, PGAP3, PGK1, PGM1, PGM3, PHEX, PHF6, PHF8, PHGDH, PHKG2, PHYH, PI4KA, PIBF1, PIEZO1, PIEZO2, PIGA, PIGB, PIGG, PIGK, PIGL, PIGN, PIGO, PIGP, PIGQ, PIGS, PIGT, PIGV, PIGY, PIK3CD, PIK3R1, PIP5K1C, PISD, PITX3, PJVK, PKD1L1, PKHD1, PKLR, PLA2G6, PLAA, PLCB1, PLCB4, PLCE1, PLEC, PLEKHG2, PLEKHG5, PLG, PLK4, PLOD1, PLOD2, PLOD3, PLP1, PLPBP, PLS3, PLVAP, PMM2, PMP22, PMPCA, PMPCB, PNKP, PNP, PNPLA1, PNPLA6, PNPLA8, PNPO, PNPT1, POC1A, POC1B, POLA1, POLE, POLG, POLG2, POLR1C, POLR1D, POLR3A, POLR3B, POMC, POMGNT1, POMGNT2, POMK, POMP, POMT1, POMT2, POP1, POR, PORCN, POU1F1, POU3F4, PPA2, PPIB, PPIP5K2, PPP1R15B, PPP1R21, PPT1, PQBP1, PRDM12, PRDM5, PRDX1, PREPL, PRF1, PRG4, PRICKLE1, PRKCD, PRKDC, PRKRA, PRMT7, PROC, PRODH, PROP1, PROS1, PRPS1, PRRX1, PRSS12, PRSS56, PRUNE1, PRX, PSAP, PSAT1, PSMB8, PSPH, PTCHD1, PTF1A, PTH1R, PTPN14, PTPN23, PTPRC, PTPRQ, PTRH2, PTS, PUS1, PUS7, PXDN, PYCR1, PYCR2, PYGL, PYGM, PYROXD1, QARS1, QDPR, RAB18, RAB23, RAB27A, RAB33B, RAB39B, RAB3GAP1, RAB3GAP2, RAC2, RAD21, RAD50, RAD51C, RAG1, RAG2, RALGAPA1, RAPSN, RARB, RARS1, RARS2, RAX, RBBP8, RBCK1, RBM10, RBM8A, RDH11, RDX, RECQL4, RELN, REN, RETREG1, RFT1, RFX5, RFX6, RFXANK, RFXAP, RIC1, RIMS2, RIN2, RINT1, RIPK1, RIPK4, RIPOR2, RLIM, RMND1, RMRP, RNASEH2A, RNASEH2B, RNASEH2C, RNASET2, RNF113A, RNF13, RNF168, RNU4ATAC, ROBO3, ROGDI, ROR1, ROR2, RPE65, RPGRIP1, RPGRIP1L, RPIA, RPL10, RPS6KA3, RRM2B, RSPH1, RSPH3, RSPO2, RSPO4, RSPRY1, RTEL1, RTN4IP1, RTTN, RUSC2, RXYLT1, RYR1, S1PR2, SACS, SAMD9, SAMHD1, SAR1B, SARS2, SASS6, SBDS, SBF1, SBF2, SC5D, SCAPER, SCARB2, SCARF2, SCN1B, SCN4A, SCN9A, SCNN1A, SCNN1B, SCNN1G, SCO1, SCO2, SCYL1, SCYL2, SDCCAG8, SDHA, SDHAF1, SDHD, SEC23A, SEC23B, SEC24D, SELENOI, SELENON, SEPSECS, SERAC1, SERPINB6, SERPINF1, SERPINH1, SETX, SFTPB, SFXN4, SGCA, SGCB, SGCD, SGCG, SGO1, SGPL1, SGSH, SH2D1A, SH3PXD2B, SH3TC2, SHOX, SHROOM4, SIL1, SKIC2, SKIC3, SLC10A7, SLC12A1, SLC12A3, SLC12A5, SLC12A6, SLC13A5, SLC16A1, SLC16A2, SLC17A5, SLC18A3, SLC19A2, SLC19A3, SLC1A4, SLC22A5, SLC25A1, SLC25A12, SLC25A13, SLC25A15, SLC25A19, SLC25A20, SLC25A22, SLC25A26, SLC25A3, SLC25A38, SLC25A4, SLC25A42, SLC25A46, SLC26A2, SLC26A3, SLC26A4, SLC26A5, SLC27A4, SLC29A3, SLC2A1, SLC2A10, SLC2A2, SLC30A10, SLC33A1, SLC34A1, SLC34A3, SLC35A1, SLC35A2, SLC35A3, SLC35C1, SLC35D1, SLC37A4, SLC39A13, SLC39A14, SLC39A4, SLC39A8, SLC3A1, SLC46A1, SLC4A1, SLC4A4, SLC52A2, SLC52A3, SLC5A1, SLC5A5, SLC5A6, SLC5A7, SLC6A3, SLC6A5, SLC6A8, SLC6A9, SLC7A7, SLC9A1, SLC9A3, SLC9A6, SLX4, SMAD4, SMARCAL1, SMC1A, SMOC1, SMPD1, SMPD4, SMS, SNAP29, SNORD118, SNX10, SNX14, SOD1, SOST, SOX3, SP110, SP7, SPAG1, SPARC, SPART, SPATA5, SPEG, SPG11, SPINK5, SPINT2, SPR, SPTBN2, SPTBN4, SQSTM1, SRD5A2, SRD5A3, SSR4, ST14, ST3GAL3, ST3GAL5, STAC3, STAG2, STAMBP, STAR, STAT1, STAT2, STAT5B, STIL, STIM1, STN1, STRA6, STRADA, STS, STT3A, STUB1, STX11, STXBP2, SUCLA2, SUCLG1, SUFU, SUMF1, SUOX, SURF1, SVBP, SYN1, SYNE1, SYNE4, SYNJ1, SYP, SZT2, TAC3, TACO1, TACR3, TAF1, TAF13, TAF2, TAF6, TAFAZZIN, TALDO1, TANGO2, TAP1, TAPT1, TARS2, TASP1, TAT, TBC1D20, TBC1D23, TBC1D24, TBC1D8B, TBCD, TBCE, TBCK, TBX15, TBX19, TBX22, TBX4, TBXAS1, TCAP, TCF12, TCIRG1, TCN2, TCTN2, TCTN3, TDP2, TECPR2, TECTA, TELO2, TENM3, TENT5A, TERT, TF, TFR2, TGDS, TGFB1, TGM1, TH, THOC2, THOC6, TIMM50, TIMM8A, TIMMDC1, TJP2, TK2, TMC1, TMCO1, TMEM107, TMEM126A, TMEM126B, TMEM132E, TMEM138, TMEM165, TMEM199, TMEM216, TMEM231, TMEM237, TMEM260, TMEM38B, TMEM67, TMEM70, TMEM94, TMIE, TMPRSS3, TMPRSS6, TMTC3, TMX2, TNFRSF11A, TNFRSF11B, TNFRSF13B, TNFSF11, TNNT1, TOE1, TOP3A, TP53RK, TPI1, TPK1, TPM3, TPP1, TPRKB, TPRN, TRAF3IP1, TRAIP, TRAK1, TRAPPC11, TRAPPC12, TRAPPC2, TRAPPC4, TRAPPC9, TRDN, TREX1, TRIM2, TRIM32, TRIM37, TRIOBP, TRIP11, TRIP13, TRIP4, TRIT1, TRMT1, TRMT10A, TRMT10C, TRMT5, TRMU, TRNT1, TRPM6, TRPV6, TSEN15, TSEN2, TSEN54, TSFM, TSHB, TSHR, TSPAN7, TSPEAR, TSPYL1, TTC19, TTC21B, TTC26, TTC7A, TTC8, TTI2, TTN, TTPA, TUBGCP2, TUBGCP4, TUBGCP6, TUFM, TUSC3, TWIST2, TWNK, TXN2, TXNDC15, TXNL4A, TYK2, TYMP, TYR, TYRP1, UBA1, UBA5, UBE2A, UBE2T, UBE3B, UBR1, UCHL1, UFC1, UFM1, UGDH, UGP2, UGT1A1, UMPS, UNC13D, UNC80, UPB1, UPF3B, UQCC2, UQCRB, UQCRC2, UQCRFS1, UQCRQ, UROC1, UROS, USB1, USH1C, USH1G, USH2A, USP18, USP53, USP9X, UVSSA, VAC14, VAMP1, VARS1, VARS2, VDR, VIPAS39, VLDLR, VMA21, VPS11, VPS13B, VPS13D, VPS33A, VPS33B, VPS37A, VPS41, VPS45, VPS51, VPS53, VRK1, VSX2, WARS2, WAS, WASHC5, WBP2, WDPCP, WDR19, WDR35, WDR4, WDR45, WDR45B, WDR62, WDR73, WDR81, WFS1, WHRN, WNK1, WNT1, WNT10A, WNT10B, WNT2B, WNT3, WNT4, WNT7A, WRAP53, WRN, WWOX, XIAP, XPA, XPC, XRCC2, XRCC4, XYLT1, XYLT2, YARS2, YIF1B, ZAP70, ZBTB24, ZC3H14, ZC4H2, ZDHHC9, ZFYVE26, ZIC3, ZMPSTE24, ZNF335, ZNF711, ZNHIT3